The phenomenon of chemical
ionization was
first observed by Munson and Field [4] in 1966. If
a large excess of a reagent gas is employed together with the sample
(the partial pressure of the reagent gas is arranged to be
about two orders of magnitude greater than that of the sample)
an entirely different type of ionization takes place. The
procedure involves first the ionization of a reagent gas such as
methane in a simple electron impact ion source and as
there is an excess of the reagent gas, the reagent molecules are
preferentially ionized. The reagent ions then
collide with the sample molecules and produce sample + reagent ions
or in some cases protonated
ions. The process is considered to be a gentle form of
ionization, because the energy of the reagent ions never exceeds 5
electron volts including those reagent ions that are considered to
have relatively high energies.
Very little fragmentation takes place and parent ions + a proton or a
molecule of the reagent gas is produced. The molecular weight of the
parent ion is, thus, easily obtained. Little modification to the
normal electron impact source is required and a conduit for supplying
the reagent gas is all that is necessary.
The
Spectrum produced by Chemical Ionization depends strongly on the
nature of the reagent ion and, thus, different structural information
can be obtained by choosing different reagent gases. This adds
another degree of freedom in the operation of the mass spectrometer.
The reagent ion can take a number of forms. Employing methane as the
reagent ion the following reagent ions can be produced
CH4 CH4+,
CH3+,
CH2+
CH4+ + CH4 CH5+ + CH3
CH3+ + CH4 C2H4+
+ H2
Other
reactions can also occur that are not useful for ionizing the solute
molecules but, in general, these are in the minority. The interaction
of positively charged ions with the uncharged sample molecules can
also occur in a number of ways, and the four most common are as
follows:
-
Proton transfer between the sample
molecule and the reagent ion,
M
+ BH+ MH+ + B
2. There is an exchange of
charge between
the sample molecule and the reagent ion,
M
+ X+ M+ + X
-
There is simple addition of the sample
molecule to the reagent ion,
M
+ X+ MX+
-
Finally there can be anion extraction
AB
+ X+ B+
+ AX
As an example (CH5+)
ions, which are formed when methane is used as the reagent gas, will
react with a sample molecule largely by proton transfer e.g.,
M +
CH5+ MX+
+ CH4
Some reagent gases produce more
reactive
ions than others, and, consequently, produce more fragmentation.
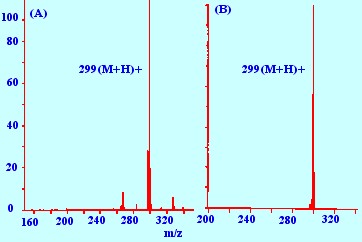
(A) Reagent Gas methane ; (B)
Reagent Gas
Isobutane.
Methane produces more active
reagent ions
than Isobutane , consequently, although methane ions produce a number
of fragments by protonation, Isobutane , by a similar protonation
process, will produce almost exclusively the protonated
molecular
ion. This is clearly demonstrated by the mass Spectrum of
methyl
stearate shown in figure 6. Spectrum (A) was produced by chemical
ionization using methane as the reagent gas and exhibits fragments
other than the protonated parent ion. In contrast, Spectrum (B)
obtained using the reagent gas butane, exhibits the protonated
molecular ion only.
The
Chemical Ionization source is very similar in design to the ion
impact source. Most mass spectrometer electron impact sources can
perform the dual role, and also act as a Chemical Ionization source.
Dual-action sources do not perform quite as well as dedicated
electron impact sources when used in the electron impact mode, but
the loss of ionization efficiency is certainly no more than 50%.
Continuous use of a source for Chemical Ionization causes significant
contamination that ultimately impairs the performance of the
spectrometer. The build-up of residues from the Chemical Ionization
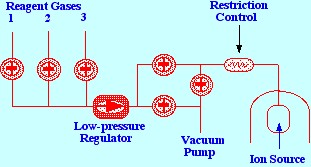
process must be regularly baked out. A diagram of a typical gas
inlet system for a Chemical Ionization source is shown in figure 7,
The
diagram depicts a system that employs three different reagent gases
but any number of reagent gases could be incorporated. The source
pressure is normally held at 0.1–0.5 torr and a low-pressure
regulator is employed to control the pressure to the required limits.
The pressure regulator and valves can be solenoid operated, and,
thus, automatically actuated by the mass spectrometer
control-computer. As a result, it is easy to change from electron
impact ionization to Chemical Ionization, as required. The sampling
procedure is relatively simple as the sample enters the mass
spectrometer as a vapor, in a gas stream, directly from the sample
vaporizer.

About the Author
RAYMOND PETER WILLIAM SCOTT was born on June 20 1924 in Erith, Kent, UK. He studied at the
University of London, obtaining his B.Sc. degree in 1946 and his D.Sc. degree in 1960.
After spending more than a decade at Benzole Producers, Ltd. Where he became head of
the Physical Chemistry Laboratory, he moved to Unilever Research Laboratories as
Manager of their Physical Chemistry department. In 1969 he became Director of Physical
Chemistry at Hoffmann-La Roche, Nutley, NJ, U.S.A. and subsequently accepted the position
of Director of the Applied Research Department at the Perkin-Elmer Corporation, Norwalk, CT, U.S.A.
In 1986 he became an independent consultant and was appointed Visiting Professor at Georgetown
University, Washington, DC, U.S.A. and at Berkbeck College of the University of London; in 1986
he retired but continues to write technical books dealing with various aspects of physical chemistry
and physical chemical techniques. Dr. Scott has authored or co-authored over 200 peer reviewed
scientific papers and authored, co-authored or edited over thirty books on various aspects of
physical and analytical chemistry. Dr. Scott was a founding member of the British chromatography
Society and received the American Chemical society Award in chromatography (1977), the
M. S. Tswett chromatography Medal (1978), the Tswett chromatography Medal U.S.S.R., (1979),
the A. J. P. Martin chromatography Award (1982) and the Royal Society of Chemistry Award in
Analysis and Instrumentation (1988).
Dr. Scott’s activities in gas chromatography started at the inception of the technique,
inventing the Heat of Combustion Detector (the precursor of the Flame Ionization Detector),
pioneered work on high sensitivity detectors, high efficiency columns and presented fundamental
treatments of the relationship between the theory and practice of the technique.
He established the viability of the moving bed continuous preparative gas chromatography,
examined both theoretically and experimentally those factors that controlled dispersion
in packed beds and helped establish the gas chromatograph as a process monitoring instrument.
Dr. Scott took and active part in the renaissance of liquid chromatography,
was involved in the development of high performance liquid chromatography and invented
the wire transport detector. He invented the liquid chromatography mass spectrometry
transport interface, introduced micro-bore liquid chromatography columns and used them
to provide columns of 750,000 theoretical plates and liquid chromatography separations
in less than a second.
Dr. Scott has always been a “hands-on” scientist with a remarkable record of accomplishments in chromatography ranging from hardware design to the development of fundamental theory. He has never shied away from questioning “conventional wisdom” and his original approach to problems has often produced significant breakthroughs.