If
any substance is exposed to radiation of a defined frequency, then
some light will be scattered at right angles. There are a number of
different types of scattering. If the excitation energy is (hν0),
and the molecule is raised from ground state to an excited level and
then falls back to the ground state, the
frequency of the light emitted will be the same as that of the
incident light and
this scattering phenomenon is called Rayleigh
scattering. If
the light is scattered by particulate
matter as by a
suspension of solids or by an emulsion the frequency of the light
emitted will also be the same as the incident light and the scattered
light but this is called Tyndale
Scattering. In
contrast, the accepted mechanism of Raman
scattering is
different to Rayleigh or Tyndale scattering but it is similar, though
not exactly the same, as Fluorescence.
In
Raman Scattering a molecule absorbs the incident radiation and, as a
consequence, is raised to a higher level of energy. The molecule then
emits light at the Raman frequency and falls to a new level, usually
somewhere between
the initial and final states.
The scattered light will contain frequencies that differ from that of the incident light and, in addition, the frequencies of
the scattered light will be characteristic
of the substance
that causes the scattering. The different forms of light scattering
are depicted diagrammatically in figure 1.
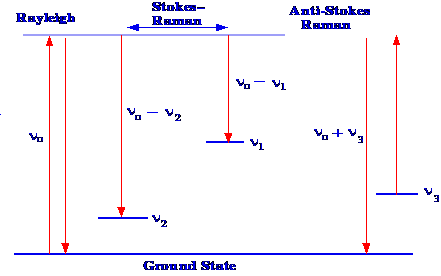
If
the frequency of the incident light is (νo)
and that of the scattered light (ν2)
then the Raman frequency
(Δν)
is given by.
Δν = ν0 - ν2(1)
If,
however, the light excites the molecule from ground level and then
falls back to an energy level of (hν1)
then the frequency of the emitted light will (ν0
–
ν1),
and this is called Stokes–Raman
scattering.
In
Stokes–Raman Scattering, the scattered light always has a
frequency lower
than that of the incident light.
Howeer,
if a molecule that already exits at a raised energy level of (hν3),
is raised further to an excited state by the incident light and then
falls back to the ground state, the frequency of the emitted light
will now be (ν0
+
ν3).
This is called Anti-Stokes-Raman
scattering, and the light scattered by this process always has a
frequency greater
than that of the incident light.
In
Anti-Stokes–Raman Scattering, the scattered light always has a
frequency higher
than that of the incident light.copy there is a mechanism by which the incident
radiation (normally its electric vector) interacts with the molecular
energy levels. Infrared adsorption is associated with molecular
vibrational levels and is detected by the changes in the dipole
moment of the molecule as it vibrates. As a consequence it is very
sensitive to polar functional groups. In contrast, Raman Spectroscopy
discloses the presence of non-polar bonds and aromatic rings. It also
discloses changes in polarization and the 'shape' of the electron
distribution in the molecules as it vibrates. The information
provided by the two techniques, Raman Spectroscopy and infrared
spectroscopy, is often claimed to be complementary. Analysis of the
Raman spectra from a wide range of compounds has shown that (hΔν)
is almost invariably equal to the change
in rotational or vibrational energy of the molecule.
If the energy levels are close to the ground state, these levels will
have a significant population as determined by the Boltzmann
distribution and, thus, be observable.
The
subject of Raman Scattering can be treated rationally by means of the
quantum theory. However the following treatment is not entirely
adequate nor is it complete but it is sufficiently so, to allow a
basic understanding of the process of Raman Scattering
Radiation of frequency (ν)
can be treated as a stream of particles having energy (hν)
where (h) is Planks constant. If the
photon striking a molecule is considered as a ball striking a solid
surface, the ball will bounce off the surface with the same energy
that it had when it struck it and, thus, the photon will have the
same energy and the scattered radiation will have the same frequency.
However, the collision may not be elastic and, thus, the photon can
gain or loose energy and the scattered light may have a greater of
lesser frequency than the incident radiation.
Thus,
if (ΔE)
is the energy change, the incident radiation has a frequency (ν)
and the scattered frequency is (ν1),
Then,
ν1
= ν ± ΔE/h
As
already discussed, if the frequency of the scattered radiation is
lower than the excitation radiation the
light it is referred to as Stokes Radiation
if the frequency is higher than the
excitation radiation it is referred to as anti-Stokes
radiation.
When
a molecule is situated in an electric field (e.g. the
undulating electric field portion of an electromagnetic wave) the
molecule becomes polarized as the positive charges in the molecule
are displaced in one direction and the negative charges in the
opposite direction giving the molecule a transient
dipole moment and, consequently, the molecule is said to be
polarized.
If
the magnitude of the induced dipole is (μ)
and the electric field (E) then,
μ
= αE
where (α)
is the polarizability of the molecule
For
radiation of frequency (ν)
the field (E) associated with it will be
given by,
E
= EoSin2πνt
Thus,
for the induced dipole. μ
= αE
= α
EoSin2πνt
This
will constitute an oscillating dipole of frequency (ν)
that itself will generate radiation also of frequency (ν),
which explains the source of Rayleigh
scattering.
Any
rotational or vibrational change in the molecule will also
periodically change the polarizability of the molecule and, thus, the
dipole will have a vibrational and/or rotational oscillation
superimposed on it.
Thus
for a vibrational frequency of (νvib)
its polarizability will be given by,
α
= αo
+ β2πνvibt
where
(αo)
is the equilibrium value of (α),
and
(β)
is the change in polarizability with external vibration.
Thus,
μ
= αE
= (αo
+ βsιν2πνvibt)Eosin2πνt
Consequently as sinAsinB
= [cos(A-B)- cos(A+B)}/2
and μ
= αo
EoSin2πνt
+ βEo
sιν(2πνvibt)sin(2πνt)
μ
= αo
EoSin2πνt
+ βEo{cos2π(ν−νvib)t-
cos2π(ν+νvib)t}/2
Consequently,
the oscillating dipole contains frequencies of ν±νvib
as well as the base
frequency
(ν).
However,
if molecular vibration and/or molecular rotations do not affect the
molecules polarizability then (β
= 0) and the scattered radiation
will have the same frequency as the exciting radiation and the
scattering will not be Raman but Rayleigh scattering.
It
should be noted that the previous statement is in contrast to the
situation with infrared and microwave absorption where molecular
motion must produce a change in the electric dipole of the molecule.
When a particular bond
between atoms produces a strong infrared signal, it is less likely to
produce a strong Raman signal, and vice
versa.
The
study of Raman spectra has a number of advantages as, by the
appropriate choice of the incident radiation, the scattered lines can
be brought into a convenient region of the Spectrum where they can be
easily observed. The energy of the incident radiation determines the
spectroscopic region where the Raman Scattering is
observed.
Originally
both incident and
scattered radiation were measured in the Visible region of the
Spectrum but, for various reasons, one of which is discussed below,
the near-infra-red radiation is now the most frequently employed.
Initially,
the practice of Raman Spectroscopy was experimentally more difficult
than today, as the intensity of the scattered light is very small
(only 0.0001% of that of the incident light, i.e.
one part in a million).
The difficulties were
greatly reduced with the
introduction of the laser light sources
of high energy in the 1960s and, in particular, the argon laser with
its intense
blue
and green
emission.
Nevertheless, although the high-intensity laser light sources has
aided in Raman spectroscopic techniques, it has also led to other
problems, such as photochemical
reaction in the
sample and sample heating with resultant black-body
radiation.
Another serious problem associated with practical Raman Spectroscopy
is the Fluorescence that can often accompany the Raman Scattering
that can be as much as six to eight orders of magnitude stronger then
the Raman light.
Often, when trying to examine Raman Scattering, Fluorescence is the only
phenomenon
observed.
The
Fluorescence can come from a variety of sources. It is often caused
by trace
impurities,
coatings on polymers ,
additives etc., that
provide so much background Fluorescence that the Raman Spectrum of
the major component cannot be discerned. The use of near
infrared excitation
can help solve this problem. It has been found, that the use of light
having a wavelength around 1 μm
to irradiate the sample virtually eliminates the fluorescent problem.
However, other problems remain such as photochemical
changes in the sample
and
blackbody
radiation
produced
by local heating.
These problems must be
carefully distinguished from Fluorescence, as the experimental
procedure necessary to correct for these effects differ greatly from
the precautions that need to be taken against Fluorescence.
The Raman Spectrum of a given substance measured
under controlled conditions can be used to confirm the identity of
the substance. An example of the different absorption curves for
infrared and Raman Spectroscopy is shown in figure 2.
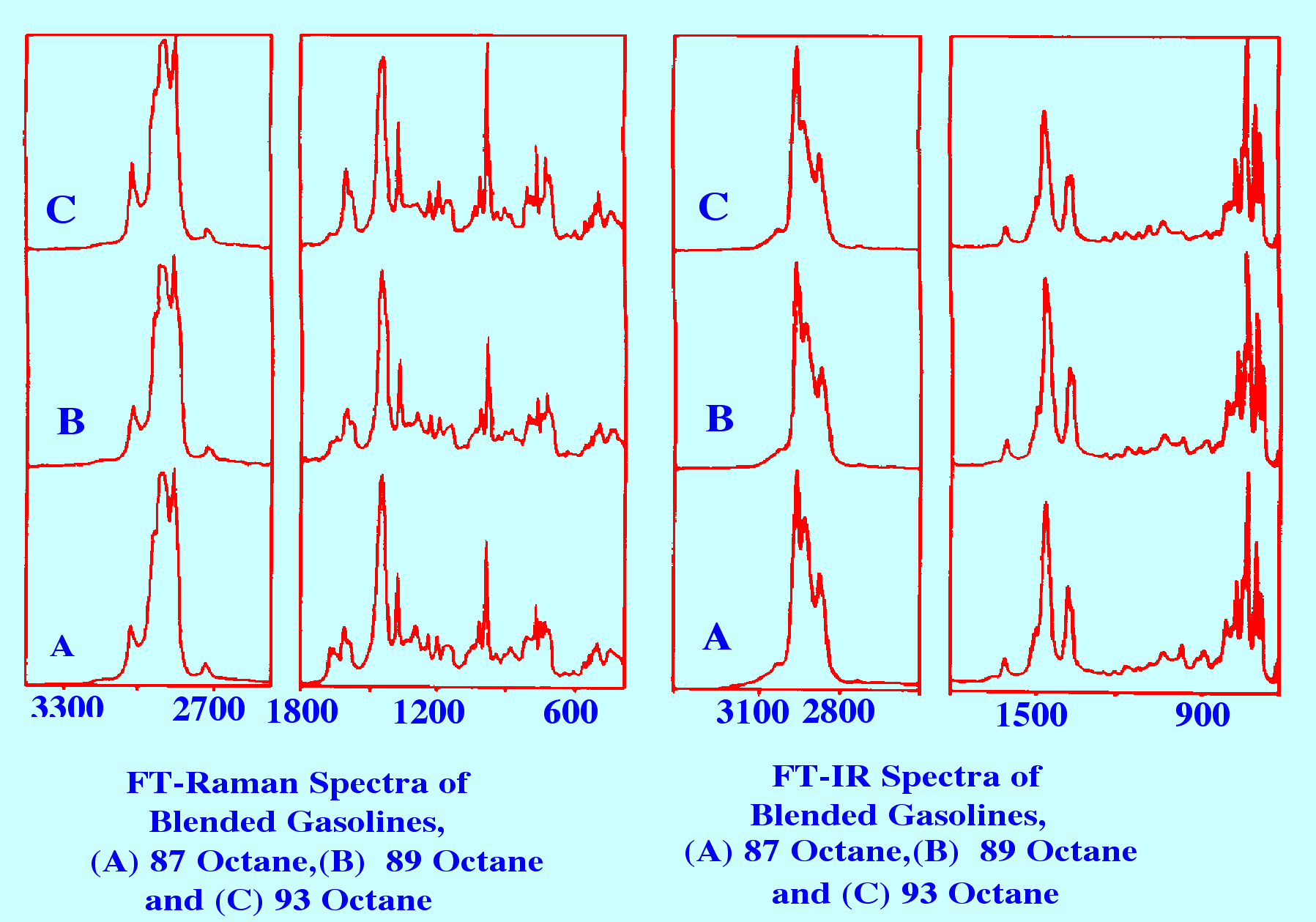
Courtesy of Nicolet Inc.
It
is seen that the spectra for the gasoline's having different Octane
rating are basically similar but, although the IR spectra show
minimal differences between the samples, there are clear and
unambiguous differences in the Raman spectra. In
figure 3 spectra are shown that have been taken from aspirin powder
by Diffuse Reflectance infrared spectroscopy and Raman Spectroscopy.
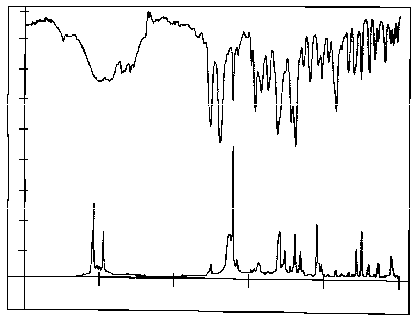
Courtesy of the Perkin Elmer Corporation
It is seen that absorbance and Raman Scattering takes place at very
similar wavelengths and there is little to choose between the two
spectra for substance identification. However if the sample is in
another form, e.g.
as the unprepared tablets themselves, the spectra differ
considerably, as shown in figure 4.
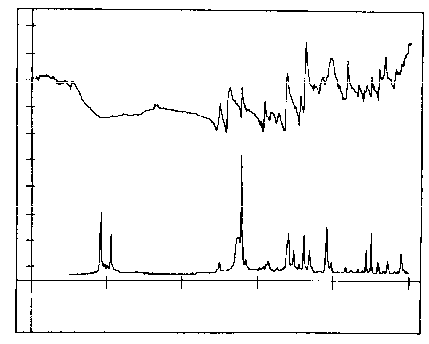
Courtesy of the Perkin Elmer Corporation
It
is seen that the spectra are now quite different;
the infrared Spectrum is virtually useless whereas the
Raman Spectrum remains similar to that obtained from the powder.
One of the great advantages of Raman Spectroscopy is that it is virtually independent of the form that the sample takes.
This could be a benefit that should be taken into account when
assessing the best spectroscopic technique to be used. Unfortunately,
the technique is even less sensitive than IR spectroscopy so its
value as an identifying technique can sometimes be rather limited.
Nevertheless, the use of a laser light source in conjunction with
FT/IR will help in this respect