Davis et al.
[18], used an
Electrospray Ionization system to couple a microbore column to a
Finnigan MAT TSQ 700 triple sector Quadrupole Mass Spectrometer. The
basic layout of the tandem system is shown in figure 27. The
microbore column was 15 cm long, 0.25 mm I.D. and packed with a C18
reversed phase support. The flow rate was only 1-2 μl
/min and, thus, a special electrospray assembly was designed to
accommodate these low flow rates. The gradient was performed and
stored in the manner of Snyder and Saunders [19] and Katz and Scott
[20]. The eluent was monitored by a UV detector, and the exit flow
from the detector passed to the micro electro-jet assembly. The
micro-spray was constructed from a flame-drawn length of fused silica
tubing, 5 cm long, 350 μm
O.D. and 150 μm I.D. The
aperture diameter of the drawn jet ranged from 1-5 μm.

Two lengths of 150 mm O.D. and
25 mm I.D.
tubing were placed inside the tube, and sandwiched between them was a
hydrophilic PVDF frit.
These insert tubes reduced the
dead volume
of the system, and provided a support for the filter, which was
essential to avoid the jet becoming blocked. Even with this
precaution, however, the tube life was limited, and could become
blocked after only a few hours of use.
The
jet was positioned accurately by means of a micrometer-driven optical
rail assembly. Spray potentials ranged from 500-1000 V, and the
optimum operating potential appeared to be about 100 V in excess of
that at which spray droplets started to form. The micro-electrospray
microbore column arrangement was used to monitor the separation of a
number of polypeptides and an example of the separation obtained form
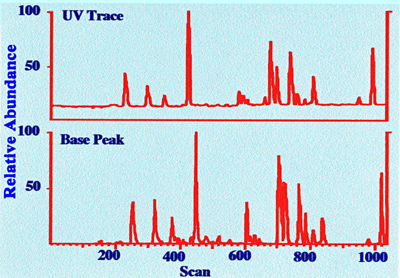
2 pmol of sample is shown in figure 28.
It
is seen the chromatograms are similar, and little or no loss of
chromatographic resolution takes place in the micro-electrospray
system. The sample size was 2 pmol (if a mean molecular weight of
1000 is assumed, this will represent a sample mass of about 2 ng).
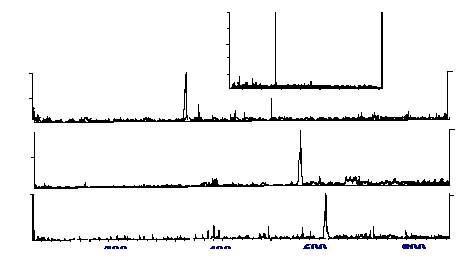
However, a better example of the sensitivity obtainable from the
system is afforded by the reconstructed chromatograms shown in figure
29. This sample size was only 40 fmole, (again assuming a mean
molecular weight of about 1000, would be equivalent to a sample mass
of 40 pg).
It
is seen that the combination of the microbore column and the
micro-electrospray provides a very high sensitivity without
compromising the performance of either the liquid chromatograph or
the mass spectrometer.
Another interesting application
not only
demonstrates the efficacy of the device for natural product
investigation, but also shows some of the foibles that can be
associated with the liquid chromatography column. Due to the increase
in contemporary popularity of herbal remedies in the United States,
there has been a need for analytical techniques to quality control
imported substances, to ensure their integrity and safety. Van
Breeman et al. [21], developed a method for
measuring the
ginsenoside content of ginseng
products, marketed as
roots, capsules, tablets and liquid extracts. ginsenoside s consist
of a series of triterpine saponins in proportions that are typical of
their country of origin. The individual ginsenoside s can be separated
by reverse phase chromatography and ion exchange chromatography, but
the use of specialized carbohydrate columns, containing aminopropyl
functional groups, have also proved useful. Van Breeman et al.,
examined the ginseng root powder by extracting the powdered root with
aqueous methanol , evaporating to dryness, dissolving the residue in
water. They extracted the ginsenoside s by passing the solution
through a solid-phase extraction tube.
A. Ginsenoside standard
added by direct infusion. B. ginsenoside standard introduced from
liquid chromatograph C. ginsenoside extract introduced from liquid
chromatograph.
The retained material was
displaced with
butanol, evaporated to dryness, and redissolved in methanol . Samples
of the methanol solution were placed on a carbohydrate analysis
column (aminopropyl bonded to silica ) and separated using a
water/acetonitrile gradient. As the ion monitored was an adduct of
sodium (M+Na)+, the mobile phase also carried
100 μ M
sodium chloride. The column eluent was monitored by a standard
Hewlett-Packard 5989B MS engine Quadrupole Mass Spectrometer, and the
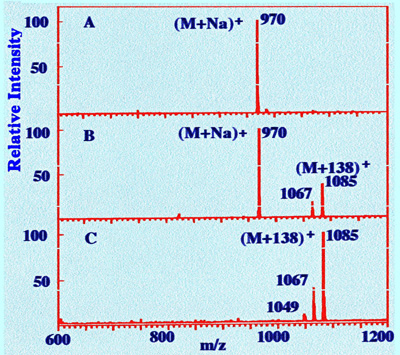
spectra that were obtained for a specific ginsenoside are shown in
figure 30.
The standard sample, injected
directly into
the mass spectrometer, gave the expected sodium adduct ion, (M+Na)+
at an m/z value of 970. However, it is seen that two new peaks
appears at m/z values of 1067 and 1085. When
the root
extract is separated and monitored under the same conditions, there
is no peak at 907 m/z and the peaks at m/z values of 1067 and 1085
have become much bigger, and that at 1085 is now the major peak.
Although all standard ginsenoside sample produced both the sodium
adduct ion and the [M+138]+ ion, the natural
products only
gave the [M+138] +.
It was also found, that the 138
adduct to
the ginsenoside was most likely to be the protonated adduct
(3-aminopropyl)-trihydroxysilane, [NH3(CH2)3Si(OH)3]+
which was present either as a reagent contaminant in the bonded phase
or was produced by the decomposition of the stationary phase. The
1067 ion appeared to be produced by the removal of water from the
[M+138]+ ion. The spectra represented only 100
pmol of
ginsenoside and so the appearance of the
(3-aminopropyl)-trihydroxysilane adducts might be a function of the
sample size and if larger charges were employed the sodium adduct
might again appear.
The size of the droplets is
vital in
electrospray techniques and Wilm and Mann [22] pointed out the lower
the flow rate the electrospray, the smaller the droplets produced.
Small droplets have high
surface to volume
ratios, and, thus, make a large proportion of the analyte molecules
available for desorption. The authors fabricated capillaries with
spraying orifices having only 1-2 μm
I.D which could accommodate flow rates as little as 20 nl/min. The
droplet diameter was claimed to be about 200 nm, in comparison with
1-2 μm, the diameter of
droplets generated from the normal electrospray sources. As a result,
the droplet volume was also 100 to 1000 times smaller and the
miniaturized electrospray inlet could be operated without a sheath
flow or pneumatic assistance, which significantly simplified the
design of the interface. The modified form of the electrospray
sampling system was not employed with an LC/MS interface, but with a
MS/MS tandem instrument. An example of a Spectrum of ovalbumin
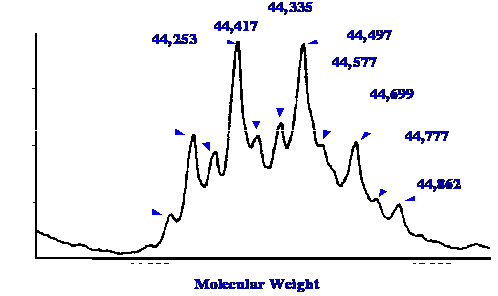
obtained using the interface is shown in figure 31.
The Spectrum was taken from a 1 μl
sample of an aqueous solution of ovalbumin containing 5 pmol/μl,
and so it was confirmed that the interface functioned well with
aqueous solvent mixtures. Assuming a molecular weight of about 44,000
for ovalbumin , this sample volume contains a mass of about 0.2 μg
of protein at a sample concentration of about 0.02%. The Spectrum is
the result of the deconvolution of the aggregate of 19 scans. The
resolution obtained at this molecular weight was about 1300.
An interesting extension of the
utilization
of this interface is its use with a MS/MS tandem instrument. The
results published by Wilm and Mann, are shown in figure 32. The top
curve shows the mass Spectrum of the peptide over the m/z range of
600 to 850. Two ion adducts from the preliminary ionization at about
m/z values 670 and 810 were chosen for subsequent examination, and
the results obtained are shown by the two spectra in the lower part
of the figure 32.
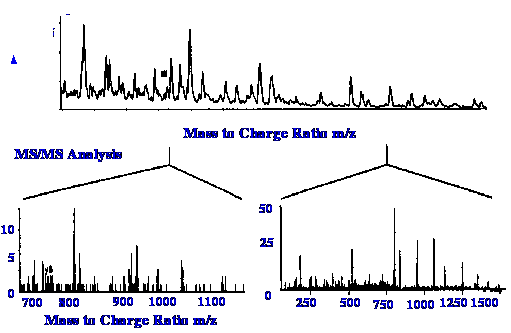
It
is seen that whereas the sample adduct ions had m/z values of 670 and
810, the m/z range of the MS/MS spectra extend to m/z values of 1100
and 1500. One explanation for this difference might be that the
original adduct ions having m/z values of 670 and 810 carried
multiple charges. It would appear that the sensitivity limits of the
electrospray interface can vary widely from one design to another,
and also, perhaps, between one type of sample and another.
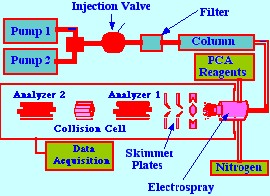
A technique using on-line
post-column
adduct formation in an electrospray inlet system was developed by
Kohler and Leary [23] for the analysis of carbohydrate mixtures. A
triaxial electrospray probe was designed that would allow the
introduction of the sample solution, the nebulizing gas and a metal
chloride reagent solution simultaneously into the probe jet. A
diagram of their apparatus is shown in figure 33.
Two LC pumps provided gradient
elution
facilities and the sample was placed on the column with a low
dispersion sample valve, having a 5μl
loop. After injection, the sample, contained in the mobile phase,
passed through a 0.5 μm
filter onto the column. A diagram of the triaxial probe is shown in
figure 34.
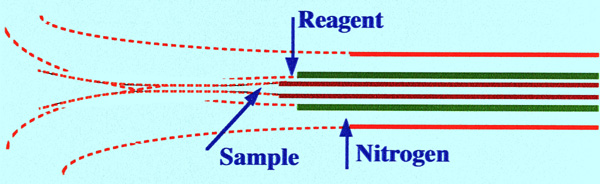
The center tube carried the
mobile phase
from the column, the next coaxial tube carried the reagent, and the
outside coaxial tube provided nitrogen to assist the nebulization.
The post-column addition of metallic chlorides to a mobile phase
carrying carbohydrates provides greater sensitivity, and assists in
structural elucidation. The relative abundance of the protonated
carbohydrate and the metal complexes with lithium , sodium , potassium ,
rubidium and cesium are shown in figure 35.
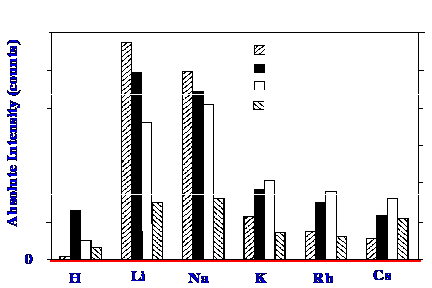
The results shown in figure 35
reveal that
the use of alkali metals complexes increases the sensitivity of the
system to the carbohydrate, resulting from the enhanced ionization in
the electrospray ionizing system. The lithium complex is shown to be
over seventy times more abundant than the protonated species.
Although the lithium complex appears to provide the maximum
sensitivity, later, but separate experiments, indicated that cobalt
complexes could give even greater sensitivity.
Employing the triaxial electrospray with
cobalt chloride as the complexing reagent, the chromatogram of 1 nmol
of a mixture of different carbohydrates, produced by single ion
monitoring, is shown in figure 36.
Although
the chromatographic separation was not complete, the single ion
monitoring of the cobalt complex ions clearly shows the presence of
each carbohydrate.
The use of different reagents to
enhance
ion production in Electrospray Ionization has been of considerable
general interest and the subject of a number of investigations It was
employed by Van Breeman [24] in the analysis of carotenoids .
Carotenoids have been shown to be the metabolic precursors of vitamin
A, and, in addition, are also thought to have anticancer activity,
and to act as in vivo antioxidants.
Van Breeman examined the use of
the
post-column injection of halogenated compounds, to enhance ionization
efficiency. The ionization efficiency of several different halogen
compounds were examined, including chloroform,
2,2,3,3,4,4,4-heptafluoro-1-butanol,
2,2,3,3,4,4,4-heptafluoro-1-butyric acid,
1,1,1,3,3,3-hexafluoro-2-propanol and trifluoroacetic acid. The
relative effect of the different additives on the efficiency of the
positive ion electrospray production was examined, and the results
that were obtained for the different halogen compounds are shown in
figure 37.

HFBA=2,2,3,3,4,4,4-heptafluoro-1-butyric
acid, TFA trifluoro acetic acid,
HFP=1,1,1,3,3,3-hexafluoro-2-propanol and
HFB=2,2,3,3,4,4,4-heptafluoro-1-butanol
The solute used in the
experiments was
β-carotene and 0.25 μg
was injected onto the column. It is seen that the presence of the
halogen antioxidants can significantly affect the ionization
efficiency. However, it is also clear that an excess of the additive
can have the opposite effect and reduces the ionization efficiency.
It is seen that the 1,1,1,3,3,3-hexafluoro-2-propanol provided the
greatest sensitivity. The limits of detection for both β-carotene
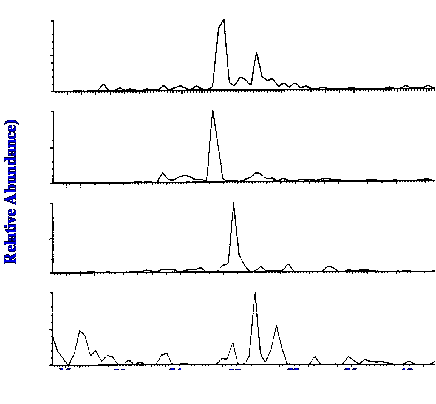
(at an m/z value of 536 and lutien (at an m/z value of 568) are shown
in figures 38 (A) and 38(B) respectively.
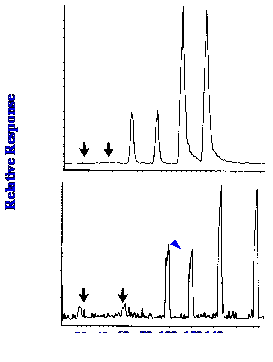
Sensitivities of about 2 pmol
and about 0.6
pmol for β-carotene and
lutein, respectively, are shown to be obtainable.
The results from the application
of the
same technique to the determination of carotenoids in heat processed
canned sweet potatoes using the reagent heptafluorobutanol is shown
in figure 39,
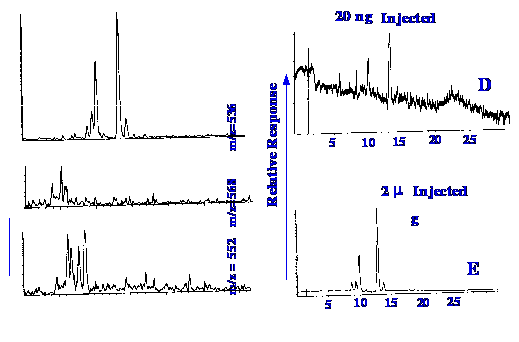
(A) to (E). (A) shows the
computer
reconstructed mass chromatogram of the β-carotene
molecular ion, at an m/z value of 536, from the injection of ca
20 ng of extract. (B) shows the computer reconstructed mass
chromatogram of the ion at an m/z value of 568, which corresponds to
lutein. (C) shows the computer reconstructed mass chromatogram of the
ion at an m/z value of 552, displying the isomers of β-cryptoxanthin.
(D) shows the chromatogram provided by the output of the Diode Array
UV detector at 450 nm, recorded during the analysis shown in (A). (E)
shows the chromatogram provided by the output of the Diode Array UV
detector at 450 nm, recorded during the separation of 2 μg
of sweet potato extract. The addition of
2,2,3,3,4,4,4-heptafluoro-1-butanol, post-column, as an ionizing
enhancer has increased the overall sensitivity to carotenoids by
about two orders of magnitude.
Sodium replacement ions can also
be used to
identify the charged state of the molecular ion. This possibility was
used by Neubauer and Anderegg [25] to identify the charged states of
peptide ions, when employing LC/MS and an electrospray ionization
system. The presence of sodium acetate at sub-millimolar levels in
the mobile phase, promotes the formation of sodium replacement ions
in addition to the normally observed protonated species. The m/z
spacing of the sodium adducts provide an unambiguous identification
of the charged state of the ions and hence their actual mass. It was
also found that at the necessary sodium concentration (ca.
250
μM) the sodium salt did
neither interfere with the chromatographic process, nor cause undue
fouling in the mass spectrometer ion source.
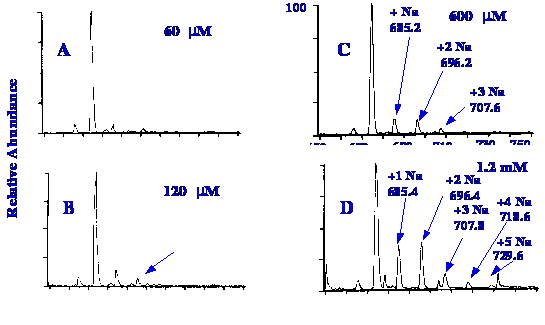
The effect of different amounts
of sodium
acetate in the mobile phase on the pattern of ion peaks produced in
the mass spectra, is shown in figure 40. It is seen that as the
quantity of sodium acetate in the mobile phase is increased, the
number of sodium replacement ions also increases. The data shown in
chromatogram (D) in figure 40 was used to identify the charge on the
molecular ion of the peptide .
The
m/z values for the five sodium replacement ions were plotted against
the number of sodium atoms in the respective replacement ion, and
curve is shown in figure 41.
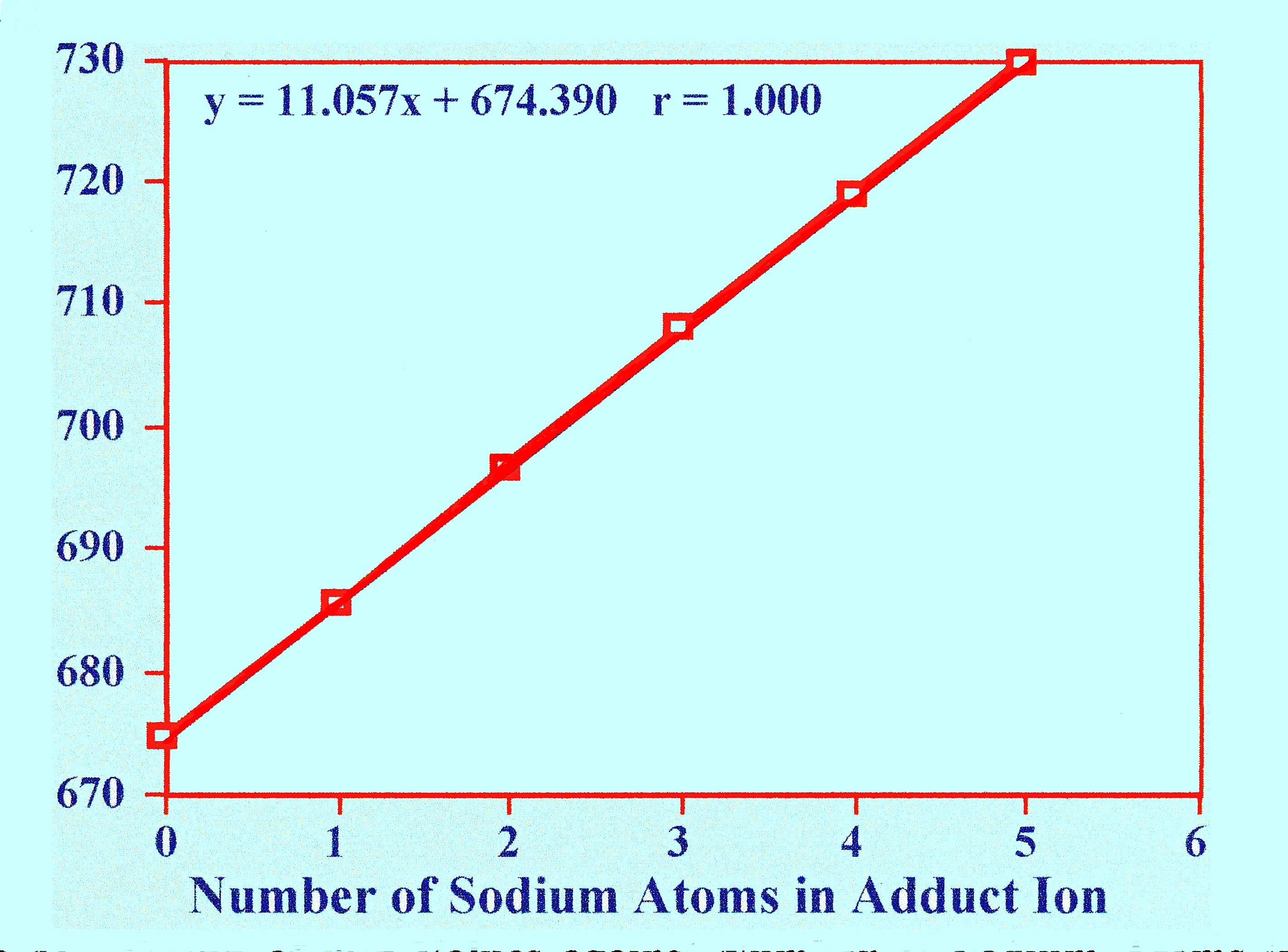
The result is a straight line,
with an
index of determination of 1.000. The slope of the line is 11.06,
which is about half the atomic weight of the sodium atom and,
consequently, the ions must be doubly charged. Taking the doubly
charged peptide ion to have m/z value of (674.6-1), (Peptide + H+),
the actual molecular weight of the peptide will probably be
(674.6-1)
x 2 =
1347.
Ionizing agents added,
pre-column and
post-column, can be used in a number of interesting and useful ways
to augment the basic information provided by the mass spectrometer.
To eliminate the restrictions
imposed by
the nature of the solvent on the function of the electrospray
ionizing system, Banks et al. [26] developed an ultrasonically
assisted nebulizer, to allow aqueous mixtures of nucleosides
to
be separated and monitored by a LC/MS tandem instrument, using the
electrospray interface. The interface they developed is shown in
figure 42.
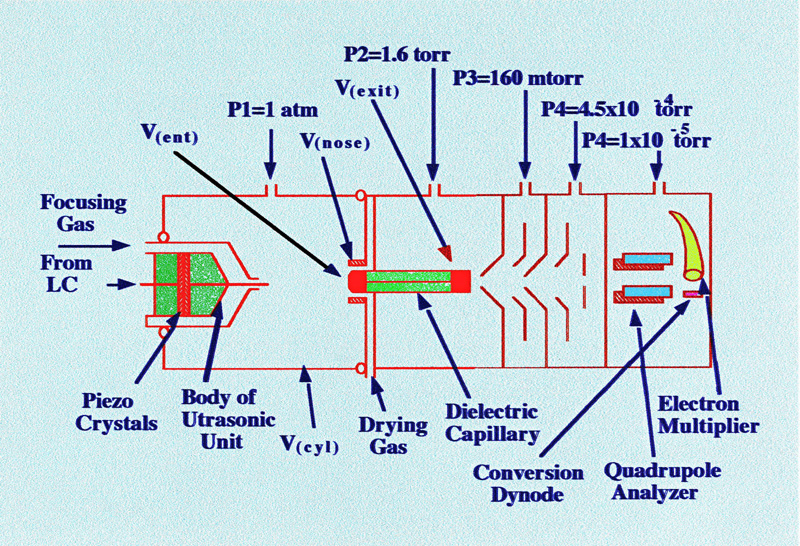
Except for the ultrasonic
nebulizing unit,
the interface was similar to those already described. It consisted of
a 0.005 in. I.D., 1/16 in. O.D., stainless steel tube, which had been
ground to a sharp point at one end, and then fitted into a two-part
stainless steel body with a nut and ferrule. A pair of piezoelectric
crystals was fitted between the two stainless steel parts, and was
driven by a function generator and a power amplifier. The device was
designed so that there was independent control of the needle
potential (V(needle)), the cylindrical electrode
potential, (V(cyl.), the nosepiece potential, (V(nose)),
and the capillary entrance potential, (V(ent)).
In the
layout shown in figure 42, the needle was always maintained at ground
potential and the temperature of the drying gas was held at 82˚C.
In other respects, the system closely resembled the standard type of
electrospray interface, typically described by Yamashita and Fenn
[27]. It was found that for effective nebulization, the ultrasonic
vibrator frequency must be adjusted to the resonant frequency of the
device, which was identified by experiment. The optimum frequency had
to be carefully controlled, as a deviation of 0.1 kHz at 180 kHz
could reduce the spray efficiency by nearly two thirds. The sample
used for optimizing the conditions of ion production was adenosine
dissolved in pure water (100 pmol/μl),
which was considered a 'worst case' example. An example of the use of
the interface in the separation of some nucleosides , using a
microbore column and a Hewlett-Packard mass spectrometer model HP88A,
is shown in figure 43.
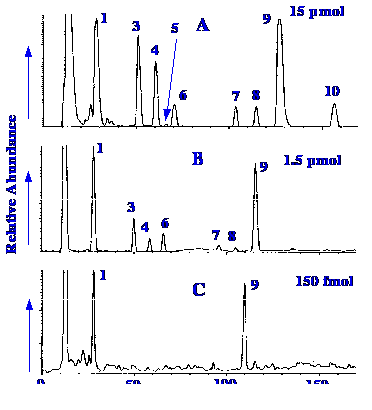
>
1. cytidine, 2. uridine, 3. 5-methylcytidine, 4. 2-O-methylcytidine, 5. 1-methyladensonine, 6. guanosine , 7. 2-O-methylguanosine,
8. N2-methylguanosine,
9. adenosine and 10.
N2-N2-dimethylguanosine.
The sample contained the
nucleosides from a
tRNA digest, reduced to its substituent nucleosides by the combined
action of nuclease P1, and bacterial alkaline phosphatase . The
sensitivity of the device is illustrated by the results shown in
figure 43. The interface operates well with liquids having high water
contents, and in fact, operates well when nebulizing samples in pure
water. It is noted, however, that very small samples exhibit
a
disproportional loss of minor components, which may make quantitative
assays uncertain when operating at maximum sensitivity. The
electrospray ionizing system has rendered the mass spectrometry
available to a wide range of applications in biology and
biochemistry, which hitherto, were precluded due to its inability of
other ionizing systems to function efficiently with aqueous
solutions.
Hua et al.
[28] employed
Electrospray Ionization in their examination of brevetoxins .
Brevetoxins are produced by the dinoflagellate , Gymnodinium
breve,
and are responsible for killing fish and also pose certain health
risks to humans. brevetoxins were isolated
from an
extract of cultured material using a reversed phase solid-state
extraction procedure. A microbore column was employed as the
separation vehicle and the eluent was split at a T-junction, part
passing to a UV detector (215nm) and the remainder to the mass
spectrometer (Vestec Model 201 fitted with an electrospray inlet
system). The Electrospray Ionization source was modified by replacing
the nozzle (the counter electrode) by a flat stainless steel plate
pierced by a 0.44 mm in diameter hole. The 200 l /min pump, normally
used to evacuate the first stage of the ion source, was replaced with
a 500 l /min. pump. These modifications reduced the lower detection
limit by a factor of four. The results obtained from a test mixture
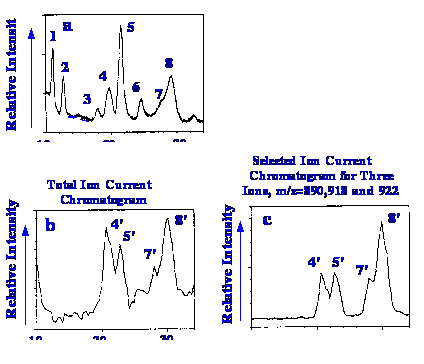
of brevetoxins are shown in figure 44. The upper chromatogram was
obtained from the UV detector and those below by total ion monitoring
and selective ion monitoring respectively. The sample consisted of 1 μl of a solution, containing
20 ng/μl (a mass of 20 ng)
and the mobile phase eluent was split in the ratio 3:1, to the UV
detector and the spectrometer respectively. The peaks correspond to
sodium adduct ions of the respective brevetoxins .
Although
the Electrospray Ionization source is one of the more popular LC/MS
interface more recently, another type of ionization source that
operates at atmospheric pressure, is now a strong competitor.